A package for coalescent analysis of patterns of linkage disequilibrium and estimation of the population recombination rate
Understanding the evolutionary forces that generate linkage disequilibrium is an important issue in both medical genetics and evolutionary biology (Pritchard and Przeworski 2001). Methods of analysis and inference based on explicit population genetic models provide powerful tools for estimating key parameters, predicting the extent of linkage disequilibrium, and testing population genetic hypotheses. LDhat is a package for analysing patterns of linkage disequilibrium within the framework of coalescent theory - a statistical description of the genealogical history of a sample of genes (Hudson 1991).
In simple population genetics models the key parameter in determining the extent of linkage disequilibrium is the product of the recombination rate and the effective population size, often termed the population recombination rate and written as Ner. LDhat estimates this parameter using the approximate-likelihood coalescent method of Hudson (2001). For each pair of segregating sites, the program pairwise estimates the coalescent likelihood of observing the data under a range of population recombination rates using the importance sampling method of Fearnhead and Donnelly (2001). The likelihoods are combined across pairs to provide a point estimate of the population recombination rate 4Ner. Further options enable the user to test for the presence of recombination using the test of McVean et al. (2001), and the relationship between summary statistics of linkage disequilibrium and physical distance. Output from the program can be used to investigate the goodness-of-fit of the model, plot the approximate likelihood surface of the estimate of 4Ner, and carry out additional analyses. Future improvements will implement estimation under multiple substitution models, including rate heterogeneity, and the estimation of confidence intervals by parametric bootstrap methods.
Currently, there are three programs in the package.
convert takes aligned sequences in FASTA format and outputs the files sites and locs that are needed for subsequent analyses.
pairwise estimates the population recombination rate by the method of Hudson (2001) and performs the test for recombination of McVean et al. (2001). The program requires the input of a file containing the aligned set of segregating sites and another file with the locations of the segregating sites relative to some arbitrary reference point. In the current version only sites with two alleles are analysed (SNP data). The user is prompted for a value of the population mutation rate, theta = 4Neu, for which the default value is the Watterson estimate. pairwise then estimates the coalescent likelihood of observing each pair of segregating sites, treating each pair as independent. The recombination rate for the entire region is then estimated over a grid with size and spacing defined by the user. A number of output files are generated: outfile contains the estimated approximate likelihood surface, fit contains the difference in log-likelihood between the best-fitting model and the ML model for each pair of segregating sites, and new_lk is the estimated likelihood surface. If the option to test for recombination is invoked, the file rdist is produced which gives the results for each permuted data set. The likelihood file, new_lk, can be used in subsequent analyses.
permute can be used for further analysis of the relationship between summary statistics of linkage disequlibrium and physical distance. As with pairwise, the sites and locs files are required, and there is an option to include the likelihood file generated by pairwise. There are three options, (1) Analyse all pairs of segregating sites; (2) Analyse pairs which are informative about recombination and (3) Analyse all sites which have a rare allele greater than some defined cut-off frequency [0-1]. Options (1) and (3) are self-explanatory. Option (2) relates to the analysis of McVean (2001) which shows that differences in the conclusions about the relationship of linkage disequilibrium to physical distance resulting from analysing different summary statistics (r2 or |D'|) can be reduced by just considering pairs of segregating sites that are informative about recombination. This condition can be obtained by considering the ratio of the log likelihood of observing the data with no recombination, to the log likelihood of observing the data for some high recombination rate. The cut-off log-likelihood ratio is arbitrarily set at 1.0, though this can be changed.
The example data (sequences and locations) set included is the LPL Jackson data from 24 African Americans collected and analysed by Nickerson et al. (1998). Diploid resequencing was carried out on a 9.7kb region from 24 individuals and phase was inferred by the method of Clark (1990). Among the 48 sequences, a total of 78 segregating sites were found, for which the Watteron estimate of theta per site is 0.0018. This value was used to estimate the likelihood of observing each pair of informative segregating sites (exclusing indel polymorphisms and microsatellites) for a range of population recombination rates from 0 to 100. The approximate likelihood surface for the total sequence is obtained by summing log-likelihoods for each pair of sites.
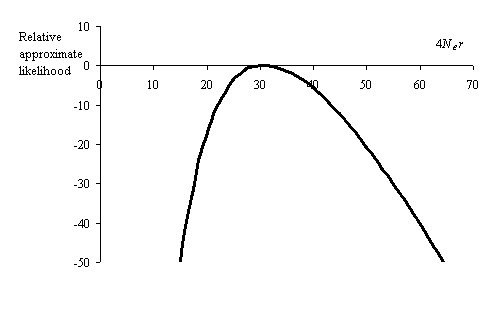
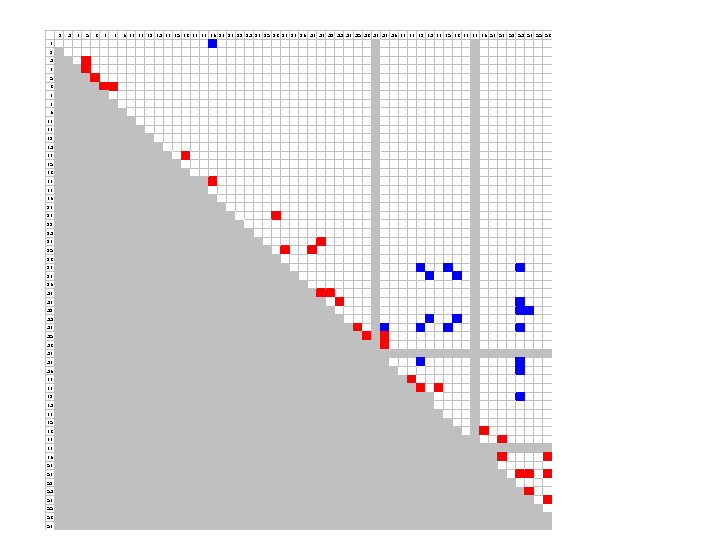
For this data set, the point estimate of the population recombination rate is 4Ner = 31. The fit between model and data can be investigated by comparing the likelihood for each pairwise comparison at the fitted recombination rate with the maximum likelihood value. Standard graphing and statistical packages such as SPlus and Excel can be used to identify pairs for which there is a significant difference between fitted and ML models. An example for the LPL data is given below. Pairs for which there is an apparent excess of LD relative to the fitted model are coloured blue (difference in log-likelihood greater than 2 units), and those showing too little LD for the fitted value are in red. Grey squares indicate sites not variable in the population.
The package can be downloaded either as source code for UNIX/Linux machines (with makefile), or as precompiled executables for DOS/Windows. Likelihood estimation within the coalescent framework uses a computationally intensive method developed by Fearnhead and Donnelly (2001) - see http://www.stats.ox.ac.uk/~fhead. For this reason, analyses of large data sets can last several hours, or even days, depending on the machine, and there is a limit of 100 sequences currently imposed.
See the README file for more details. The archived package also contains an example data set for which the likelihood file can be downloaded here.
Fearnhead, P. and Donnelly, P. (2001). Estimating recombination rates from population
genetic data.; Genetics in press (download
ps file)
Hudson, R. R. (1991) Gene genealogies and the coalescent process. In Oxford Surveys in Evolutionary Biology (Eds D. Futuyama and J. Antonovics), volume 7, pp. 1-44. Oxford University Press.
Hudson, R. R. (2001) Two-locus sampling distributions and their application. Genetics in press
(Hudson's home
page)
McVean, G. A. T. (2001) What do patterns of genetic variability reveal about mitochondrial recombination? Heredity, in press (download pdf file).
McVean, G. A. T., Awadalla, P. and Fearnhead, P. (2001). A coalescent-based method for detecting recombination from gene sequences. In preparation. (download pdf file)
Pritchard, J. K. and Przeworski, M. (2001). Linkage disequilibrium in humans: models and data. Am. J. Hum. Genet. 69: 1-14. (download pdf file).
Nickerson, D. A. et al. (1998) DNA sequence diversity in a 9.7-kb region of the human lipoprotein lipase gene. Nature Genet.19: 233-240.
Last update: 7/9/01